
โรคปัสสาวะกลิ่นเมเปิลไซรับ
เรียบเรียงโดย นายธีรพัฒน์ เวชชประสิทธิ์
โรคปัสสาวะกลิ่นเมเปิลไซรับ (Maple syrup urine disease; MSUD) หรือ โรคฉี่หอม ที่ได้ชื่อโรคเช่นนี้เนื่องจากปัสสาวะของผู้ที่ป่วยจะมีกลิ่นคล้ายกับกลิ่นของเมเปิลไซรับ
ผู้ป่วย MSUD จะเริ่มแสดงอาการตั้งแต่อยู่ในวัยทารก โดยเมื่อทารกแรกเกิดได้รับสารอาหารประเภทโปรตีน เช่น น้ำนมจากแม่ โปรตีนส่วนที่เกินความจำเป็นที่ร่างกายจะนำไปใช้จะส่งผลให้เกิดอันตรายต่อทารก ทำให้มีอาการเบื่ออาหาร ดูดน้ำนมได้น้อย น้ำหนักตัวลดลง ร้องไห้เสียงแหลมสูงเป็นช่วงสั้น (high pitched cry) และปัสสาวะมีกลิ่นคล้ายเมเปิลไซรับ จากนั้นจะเกิดความผิดปกติที่เรียกว่า metabolic crisis โดยทารกจะแสดงอาการง่วงนอน เกียจคร้าน มีอารมณ์แปรปรวน ร่วมกับการอาเจียน หากไม่ได้รับการรักษาอาจเกิดภาวะแทรกซ้อนอื่นๆ ตามมาอีก เช่น ชัก เกิดภาวะสมองบวม หรือมีภาวะความเป็นกรดในเลือดสูง ซึ่งบางรายอาจมีอาการตาบอด สมองถูกทำลาย และอาการอาจรุนแรงจนถึงขั้นเสียชีวิตได้ ผู้ป่วยที่ไม่ได้รับการตรวจรักษาจึงมักมีชีวิตอยู่ได้ไม่กี่เดือนเท่านั้น
จากผลการตรวจคัดกรองเด็กแรกเกิดทั่วโลกจำนวน 26.8 ล้านคน พบเด็กที่ป่วยเป็น MSUD ประมาณ 1 ใน 185,000 คน ในประเทศไทยโรค MSUD จะถูกตรวจพบเมื่อผู้ป่วยแสดงอาการของโรคออกมาแล้ว ทั้งนี้เนื่องจาก MSUD ไม่ใช่โรคสำคัญในการตรวจคัดกรองโรคของทารกแรกเกิด รวมทั้งโรงพยาบาลในส่วนภูมิภาคส่วนใหญ่ขาดเครื่องมือที่ใช้สำหรับตรวจวินิจฉัยโรคชนิดนี้
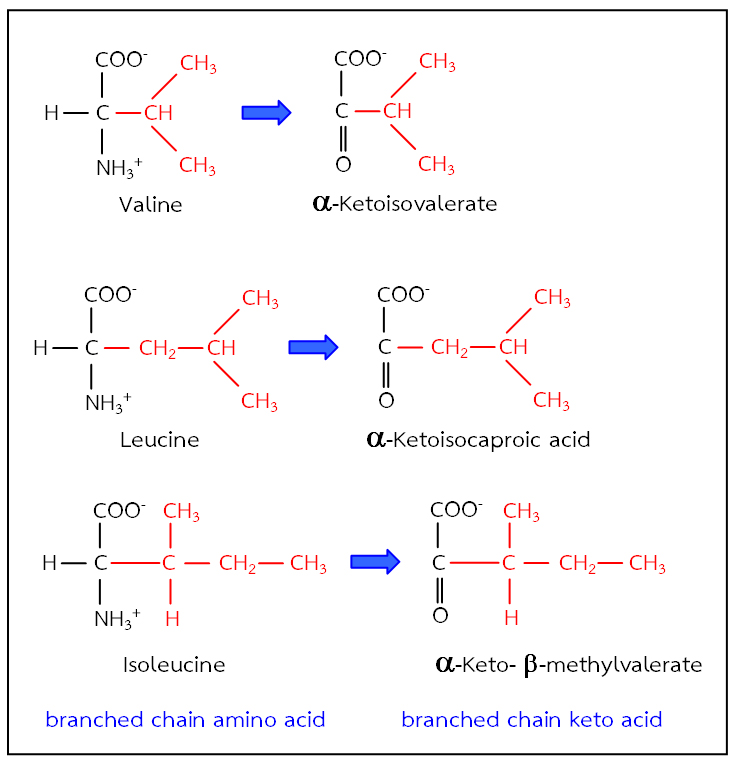
ภาพที่ 1 โครงสร้างของ branched chain acid และ branched chain keto acid
MSUD เป็นโรคที่เกิดจากความผิดปกติของเมแทบอลิซึม เนื่องจากร่างกายของผู้ป่วย MSUD ไม่สามารถสลายกรดอะมิโน 3 ชนิด คือ วาลีน (valine) ลิวซีน (leucine) และไอโซลิวซีน (isoleucine) โดยการสะสมของกรดอะมิโนดังกล่าวจะมีผลต่อสมอง และทำให้เกิดกลิ่นหอมคล้ายเมเปิลไซรับปนออกมากับปัสสาวะของผู้ป่วย
กรดอะมิโนชนิดต่างๆ ที่อยู่ในร่างกายจะถูกนำมาใช้ในการสร้างโปรตีน แต่เมื่อมีการสะสมของกรดอะมิโนเกิดขึ้น ร่างกายจะมีกลไกการเปลี่ยนกรดอะมิโนเหล่านี้ให้อยู่ในรูปของคาร์บอนและไนโตรเจน คาร์บอนที่ได้จะถูกนำไปใช้ในการสร้างกลูโคสหรือกรดไขมันชนิดต่างๆ ในขณะที่ไนโตรเจนจะถูกนำกลับมาใช้ใหม่ หรือไนโตรเจนส่วนเกินจะถูกกำจัดออกในรูปของยูเรีย
กรดอะมิโนลิวซีน ไอโซลิวซีน และวาลีน เป็นกรดอะมิโนที่มีโครงสร้างแบบกิ่งก้าน (branched chain amino acid) ซึ่งในคนปกติร่างกายจะสลายกรดอะมิโนทั้ง 3 ชนิดนี้ โดยกลุ่มเอนไซม์ branched chain amino acid transaminase ให้อยู่ในรูปของ branched chain keto acid ดังภาพที่ 1 จากนั้น branched chain keto acid เหล่านี้จะถูกสลายโดยกลุ่มเอนไซม์ที่มีชื่อว่า branched chain alpha-keto acid dehydrogenase; BCKD จนได้ผลิตภัณฑ์สุดท้ายเป็น กรดอะซิโตแอซิติก (acetoacetic acid) แอซิทิลโคเอนไซม์เอ (acetyl-CoA) หรือ ซัคซินิลโคเอนไซม์เอ (succinyl-CoA) แต่ในผู้ป่วย MSUD จะมีความผิดปกติของกลุ่มเอนไซม์ BCKD ทำให้การ สลายกรดอะมิโนทั้ง 3 ชนิดนี้ทำได้ไม่สมบูรณ์
กลุ่มเอนไซม์ BCKD อยู่บริเวณเยื่อหุ้มชั้นในของไมโทรคอนเดรีย ประกอบด้วยเอนไซม์ที่ทำหน้าที่เร่งปฏิกิริยา 3 ชนิดด้วยกัน คือ ดีคาร์บอกซิเลส (decarboxylase; E1) ทรานแอซิลเลส(transacylase; E2) และ ดีไฮโดรจีเนส (dehydrogenase; E3) ดังภาพที่ 2 ความผิดปกติที่เกิดขึ้นกับยีนต่างๆ ที่อยู่บนออโตโซมซึ่งควบคุมการสร้างเอนไซม์ E1 E2 หรือ E3 นี้เป็นสาเหตุของการเกิด MSUD
การสร้างเอนไซม์ E1 ซึ่งประกอบด้วยโปรตีนหน่วยย่อย 2 ชนิดคือ หน่วยย่อยอัลฟา (alpha subunit) และหน่วยย่อยบีตา (beta subunit)ควบคุมโดยยีน 2 ชนิดคือ “branched chain keto acid dehydrogenase, alpha polypeptide; BCKDHA” ซึ่งอยู่บนแขนข้างยาวของโครโมโซมแท่งที่ 19 ยีนนี้ควบคุมการสร้างโปรตีนที่เป็นส่วนประกอบของหน่วยย่อยอัลฟาของเอนไซม์ E1 และยีน “branched chain keto acid dehydrogenase, beta polypeptide; BCKDHB” ซึ่งอยู่บนแขนข้างยาวของโครโมโซมแท่งที่ 6 ยีนนี้ควบคุมการสร้างโปรตีนที่เป็นส่วนประกอบของหน่วยย่อยบีตาของเอนไซม์ E1 ความผิดปกติที่เกิดขึ้นกับยีนทั้ง 2 ชนิดนี้เป็นผลทำให้เกิดการเปลี่ยนแปลงของกรดอะมิโน 1 ชนิดในโครงสร้างของหน่วยย่อยอัลฟาและบีตาของเอนไซม์ E1
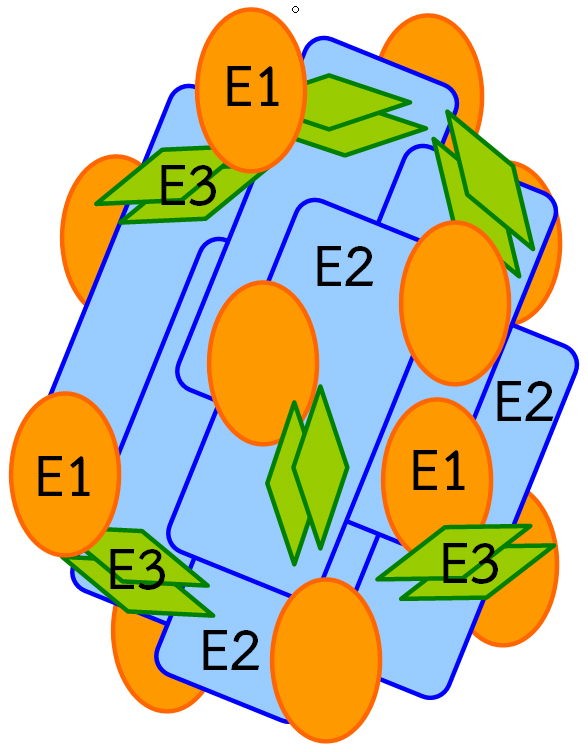
ภาพที่ 2 กลุ่มเอนไซม์ BCKD
การสร้างเอนไซม์ E2 ควบคุมโดยยีน “dihydrolipoamide branched chain transacylase; DBT” ซึ่งอยู่บนแขนข้างสั้นของโครโมโซมแท่งที่ 1 ความผิดปกติของยีนนี้เกิดจากการเปลี่ยนแปลงชนิดของเบสตัวใดตัวหนึ่งบนสาย DNA รวมทั้งการเพิ่มหรือลดชิ้นส่วนของ DNA บริเวณยีนนี้ ส่วนการสร้างเอนไซม์ E3 ควบคุมโดยยีน “dihydrolipoamide dehydrogenase;DLD” ซึ่งอยู่บนแขนข้างยาวของโครโมโซมแท่งที่ 7 ความผิดปกติของยีนนี้ทำให้เกิดการเปลี่ยนแปลงของกรดอะมิโน 1 ชนิดในโครงสร้างของเอนไซม์ E3
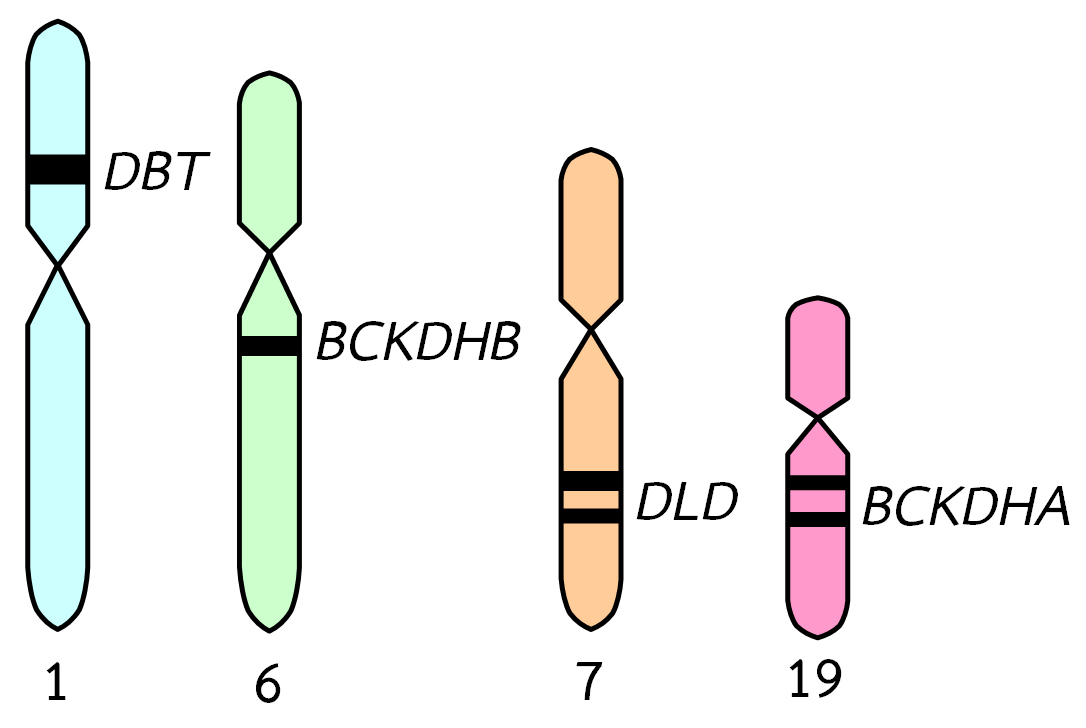
ภาพที่ 3 ยีนควบคุมการสร้างเอนไซม์ E1 E2 และ E3 บนโครโมโซมแท่งต่างๆ
แม้จะมีงานวิจัยว่า MSUD สามารถรักษาให้หายได้โดยการผ่าตัดเปลี่ยนตับจากคนปกติ แต่ผู้ป่วย MSUD ที่ไม่ได้รับการผ่าตัดเปลี่ยนตับใหม่ก็สามารถดำรงชีวิตแบบปกติได้จนเป็นผู้ใหญ่ได้ แต่ต้องได้รับการดูแลจากแพทย์และผู้เชี่ยวชาญทางด้านโภชนาการไปตลอดชีวิต โดยเฉพาะในทารกแรกเกิดต้องได้รับอาหารที่มีปริมาณโปรตีนพอเพียงกับที่ร่างกายต้องการเท่านั้นและต้องได้รับโดยทันทีหลังจากที่มีการตรวจพบว่าทารกป่วยเป็นโรค MSUD

ภาีพที่ 4 นมสูตรเฉพาะของทารกที่ป่วยเป็น MSUD
ผู้ป่วย MSUD นอกจากต้องได้รับอาหารที่เป็นสูตรเฉพาะที่มีการควบคุมปริมาณโปรตีนแล้ว ยังต้องหลีกเลี่ยงอาหารชนิดต่างๆ ที่มีปริมาณกรดอะมิโนโครงสร้างแบบกิ่งก้านสูงๆ เช่น น้ำนมวัว เนื้อสัตว์ ปลา เนยแข็ง ไข่ รวมทั้งอาหารประเภทแป้งและถั่วเมล็ดแห้งต่างๆ ส่วนอาหารที่มีปริมาณกรดอะมิโนโครงสร้างแบบกิ่งก้านอยู่น้อยเช่น ผักและผลไม้ชนิดต่างๆ สามารถรับประทานได้แต่ก็จำเป็นต้องควบคุมให้อยู่ในปริมาณที่เหมาะสม เพื่อป้องกันไม่ให้มีความผิดปกติเกิดขึ้นแม้จะไม่ได้อยู่ในวัยทารกแล้วก็ตาม
เอกสารอ้างอิง
1. Dean, j. D. and Christopher, B. D. Human mutations affecting branched chain a-ketoacid dehydrogenase. Frontiers in bioscience 3. 517-524. 1998
2. Donald, V. and Judith, G. V. Biochemistry. John Wiley & Sons, Inc. USA. 1990
3. Suthipong, P., Wiyada, C. and Varaporn S. Maple syrup urine disease in Thai infants. J. Med Assoc Thai vol. 91. 41-44. 2008
4. Genetic fact sheets for parents amino acid disorders : Maple syrup urine disease MSUD type 1A.http://www.newbornscreening.info/Parents/aminoaciddisorders/MSUD.html retrieved 7 ธ.ค.54
5. BCKDHA, BCKDHB, DBT and DLD. http://ghr.nlm.nih.gov/BrowseGenes retrieved 29 ธ.ค. 54
6. Liver transplantation for classical maple syrup urinedisease. http://www.sciencedirect.com/science/article/pii/S0022347611006603 retrieved 29 ธ.ค. 54
7. ที่มาภาพที่ 4 http://www.cardinal.com/us/en/distributedproducts/ASP/MJ041802.asp?cat=med_surg
22,414 total views, 2 views today